Monoclonal antibodies are a group of biologic medications that have been marketed in the United States for cancer treatment since 1997.1 Since then, the list of antibodies approved by the US Food and Drug Administration (FDA) has risen dramatically, with approximately 50% of new approvals or expanded indications in May 2021 to May 2022 incorporating a monoclonal antibody.2 With this rise in approvals and expanded indications for monoclonal antibodies comes an increase in overall drug spending, with monoclonal antibodies being the most costly drug class.1 Thus, there is a considerable effort to mitigate the exponential costs that can arise with the increased use of increasingly costly drugs.
The Drug Price Competition and Patent Term Restoration Act of 1984, or the Hatch-Waxman Amendment, offered a regulatory pathway for generic small-molecule medications to be approved by the FDA; however, this did not offer any pathway for biologics.1 Because the portfolio of available agents was minimal during that time, it made sense to not include biologics. However, with the rise of biologic agents, a novel method for approval of what could be considered a generic drug was needed; thus, the category of biosimilar was created.
Biosimilars
Because biologic agents are more structurally complicated than small-molecule drugs, the manufacturing process of biologics involves more steps and final drugs are never identical when analyzing the entire molecule.1 Only the active sequence of amino acids that will exert the agent’s therapeutic effect is required to be identical.1 Because of this lack of identical chemical structure, biologics cannot be considered generic drugs as typically thought of with small-molecule medications. Rather, these agents are defined as highly similar or biosimilar.3
The legal pathway for biosimilars arose with the passage of the Biologics Price Competition and Innovation Act (BPCIA), an amendment to the Public Health Service Act that was signed into law as part of the Affordable Care Act.4 The BPCIA created the 351(k) approval pathway for biosimilars if they are “highly similar” to the reference drug, except for small changes in clinically inactive components of the molecule.4 Through an abbreviated approval pathway, the FDA requires biosimilars to show similar pharmacokinetics, pharmacodynamics, and immunogenicity to the reference drug.3 Comparative studies that demonstrate equivalent efficacy and adverse events are not required to determine biosimilarity, although they are required to demonstrate interchangeability with the reference drug.3,4 Interchangeability would be shown through clinical efficacy and data on adverse events.3 Being interchangeable would allow for the automatic substitution of the biosimilar for the reference drug, similar to how generic, small-molecule medications can be substituted for the respective brand-name medication; however, this would still be subject to state regulations.1 To date, no biosimilars have been granted interchangeable status in oncology.
The cost of monoclonal antibodies averages slightly less than $100,000 per year, although agents used for hematology or oncology account for the most expensive drugs.5 Of all the antibodies with an average annual cost of more than $100,000, 85% are used for hematology or oncology indications. These agents have an average annual cost of $142,833, with the next most expensive group being agents used for immunology at an average annual cost of $53,969.5
Because of their immense cost, there is economic interest in adopting biosimilars among health systems, payers, and patients. Commercial payers have incorporated biosimilars into their formulary, with 67% granting a particular biosimilar preferred coverage or similar coverage to the reference drug.6 Hence, health systems also have a commercial incentive to begin the integration of biosimilars into their formularies as they are approved by the FDA. Several studies have shown a rapid adoption of filgrastim biosimilars.7,8 Filgrastim-sndz, the first filgrastim biosimilar, accounted for approximately 50% of all filgrastim drugs only 3 years after its approval, leading to an approximate 20% to 30% reduction in spending depending on the payer, including commercial payers, Medicare, and Medicaid.7,8
Currently, 5 monoclonal antibodies are approved for hematology or oncology indications that have commercially available biosimilars, including bevacizumab, filgrastim, pegfilgrastim, rituximab, and trastuzumab (epoetin alfa was not approved at the time this article was written and is therefore not included). This article reviews the available literature assessing the clinical efficacy and adverse events related to the use of these biosimilars versus using the reference drug.
Bevacizumab
Bevacizumab (Avastin) is a humanized monoclonal antibody that antagonizes vascular endothelial growth factor (VEGF), thereby inhibiting angiogenesis, which is thought to slow the growth of tissues.9 Bevacizumab has several oncologic indications, including for colorectal cancer, non–small cell lung cancer (NSCLC), renal cell carcinoma, glioblastoma, cervical cancer, ovarian cancer, and hepatocellular carcinoma. Several of the more unique adverse events associated with bevacizumab treatment include hypertension, proteinuria, bleeding, and wound dehiscence.9 There are currently 4 FDA-approved biosimilars to bevacizumab, including bevacizumab-adcd (Vegzelma), bevacizumab-awwb (Mvasi), bevacizumab-bvzr (Zirabev), and bevacizumab-maly (Alymsys).10-14
Bevacizumab-adcd was compared with reference bevacizumab in a phase 3, double-blind, multicenter study of patients with metastatic or recurrent NSCLC.15 The patients received concomitant carboplatin area under the curve (AUC) of 6.0 and paclitaxel 200 mg/m2 as induction therapy every 3 weeks for 4 to 6 weeks followed by bevacizumab-adcd (N=342) or reference bevacizumab (N=347) at a dose of 15 mg/kg every 3 weeks. The patients who had at least controlled disease after induction therapy then received bevacizumab-adcd or bevacizumab maintenance therapy.15
The patients’ baseline characteristics were similar between the 2 arms, with most patients having adenocarcinoma (98.1%).15 The primary end point of overall response rate (ORR) met the predefined noninferiority interval of –12.5% to 12.5%, with an ORR of 42.4% with the biosimilar compared with 42.07% with the reference drug (risk difference, 0.4%). Both arms had a similar number of complete responses (CRs; 0.6% vs 0.9%, respectively). There were no significant differences in the secondary end points of time to progression (8.5 months vs 8.3 months, respectively), median progression-free survival (PFS; 7.9 months vs 7.2 months, respectively), and median overall survival (OS; 17.1 months vs 15.6 months, respectively) between the biosimilar and reference bevacizumab groups. No new adverse events with biosimilar bevacizumab versus reference bevacizumab were seen, and both arms had a similar rate of adverse events, including gastrointestinal (GI) perforations (0.9% vs 1.5%, respectively) and hypertension (12.8% vs 11.3%, respectively).15
Thatcher and colleagues compared bevacizumab-awwb and bevacizumab in the phase 3 MAPLE study.16 Patients with newly diagnosed, metastatic or recurrent, nonsquamous NSCLC were randomized to receive bevacizumab-awwb (N=328) or bevacizumab (N=314) dosed at 15 mg/kg and administered in 6 cycles and carboplatin and paclitaxel administered every 3 weeks for 4 to 6 cycles. There was no maintenance phase included in this analysis. No between-group differences were seen in the patients’ baseline characteristics. The primary end point of noninferiority with ORR was met, with an ORR of 39% with bevacizumab-awwb and 41.7% with bevacizumab (relative risk [RR], 0.93; 90% confidence interval [CI], 0.8-1.09), which was within the prespecified margin of 0.67 to 1.5. The majority of ORRs in the bevacizumab-awwb and bevacizumab groups were stable disease (43.9% vs 43.6%, respectively) and partial responses (38.4% vs 41.1%, respectively). In both arms, 0.6% of the patients had a CR. There were no significant differences between the bevacizumab-awwb and bevacizumab groups in the secondary end points of median duration of response (DOR; 5.8 months vs 5.6 months, respectively) and PFS (39.9% vs 39.8%, respectively; hazard ratio [HR], 1.03; 90% CI, 0.83-1.29). There were also no significant differences in adverse events between the groups, with the most frequent being anti-VEGF adverse events (hypertension, GI perforations).16
In the phase 3, double-blind, randomized STELLA trial, bevacizumab-maly was compared with reference bevacizumab.17 Patients with newly diagnosed or recurrent stage IIIB or stage IV nonsquamous NSCLC were randomized to bevacizumab-maly (N=315) or to bevacizumab (N=312) dosed at 15 mg/kg with paclitaxel 200 mg/m2 and carboplatin AUC 6 every 3 weeks for 6 cycles followed by maintenance bevacizumab. In all, 91.9% of the patients had newly diagnosed NSCLC, and there was an even distribution of smokers and nonsmokers. The primary end point of the ORR at week 18 (end of induction) was similar between the bevacizumab-maly and the reference bevacizumab groups (40.3% vs 44.6%, respectively), with an RR of 0.91 (90% CI, 0.758-1.092), meeting the predefined noninferiority margin (0.73-1.36). Most of these responses were partial responses (38.4% vs 43.6%, respectively) or stable disease (17.1% vs 17.%, respectively). No significant differences were noted between biosimilar bevacizumab and reference bevacizumab in median PFS (36 weeks vs 37.29 weeks, respectively), median OS (not achieved at data cutoff; HR, 1.108; 95% CI, 0.83-1.49), and DOR (30.29 weeks vs 37.14 weeks, respectively; HR, 1.195; 95% CI, 0.92-1.56). There were no differences in the rates of adverse events between the 2 arms overall; however, there were slightly higher incidences of epistaxis (4.2% vs 2.9%, respectively) and hemoptysis (2.6% vs 1.3%, respectively) in the biosimilar bevacizumab arm than in the bevacizumab arm and higher incidences of proteinuria (3.9% vs 5.8%) and neutropenia (1.6% vs 3.2%, respectively) in the reference bevacizumab arm versus the bevacizumab biosimilar arm, although most of these were grade 1 or 2. There were no differences in serious adverse events between the arms.17
Based on the available data presented, there is no difference in efficacy or adverse events between bevacizumab and its biosimilars. Neither ORR, time to progression, nor survival was significantly different between bevacizumab and the bevacizumab biosimilar in any of the trials. Data from the trials of bevacizumab and its biosimilars are shown in Appendix Table 1, available Here.15-17
Filgrastim
Filgrastim (Neupogen) is a granulocyte colony-stimulating factor (G-CSF) that stimulates the production and activation of neutrophils.18 Filgrastim is indicated for the treatment of or prophylaxis for neutropenia after chemotherapy, as well as for stem-cell mobilization in patients who have had a hematopoietic cell transplant (HCT).18 Probably the most unique adverse event of treatment with filgrastim is bone and musculoskeletal pain.18 There are currently 3 FDA-approved filgrastim biosimilars, including filgrastim-aafi (Nivestym), filgrastim-ayow (Releuko), and filgrastim-sndz (Zarxio). Another agent, tbo-filgrastim (Granix), is not technically a biosimilar because a Biologics License Application was filed for this drug before the introduction of biosimilars as a regulatory pathway for approval, although tbo-filgrastim is colloquially considered a biosimilar.4,18-22
Filgrastim-aafi was assessed for noninferiority in a retrospective, 3-cohort trial that compared filgrastim (N=147), filgrastim-aafi (N=134), and pegfilgrastim (N=139) in women with early breast cancer who were receiving docetaxel, doxorubicin, and cyclophosphamide as well as prophylaxis for primary febrile neutropenia (FN).23 When comparing only the 2 filgrastim arms, more patients in the biosimilar arm were premenopausal than patients receiving the reference drug (70% vs 48%, respectively). More patients in the reference arm than in the biosimilar arm received chemotherapy in the neoadjuvant setting (100% vs 58%, respectively). Other baseline characteristics were similar between the 2 arms. There was an equal number of FN episodes (16% for both) and a similar amount of cycles with an FN episode with filgrastim-aafi and filgrastim (4% vs 3%, respectively). No significant difference was seen in documented infections in the filgrastim-aafi and filgrastim groups (14% vs 15%, respectively). However, the median absolute neutrophil count (ANC) at diagnosis of FN was lower with filgrastim-aafi than with filgrastim (95×103 cells/uL vs 200×103 cells/uL, respectively; P=.015). Patients received similar doses as well as a comparable number of doses of filgrastim or filgrastim-aafi in both arms. There were no other differences between the 2 drugs in this study. This trial did not assess for adverse events resulting from treatment with filgrastim.23
Filgrastim-sndz was compared with filgrastim in the phase 3, randomized, double-blind PIONEER trial.24 A total of 218 patients were randomized to 1 of 4 arms: filgrastim only, filgrastim-sndz only, or 1 of 2 arms in which the patients started receiving filgrastim or filgrastim-sndz and then alternated treatment (filgrastim-sndz followed by filgrastim or filgrastim followed by filgrastim-sndz) every other cycle for 6 cycles; all patients received docetaxel, doxorubicin, and cyclophosphamide. The analysis of this trial, however, only compared patients who were receiving reference filgrastim (N=52) with patients who were in one of the treatment switch arms that were combined into 1 arm (N=109).24
The patients’ baseline characteristics were similar between the 2 treatment arms, with approximately 58% of patients receiving adjuvant chemotherapy compared with approximately 42% receiving neoadjuvant chemotherapy.24 No difference was seen in the rates of FN between the drug switch and reference filgrastim arms (3.4% vs 0%, respectively), which was within the predefined noninferiority margin of –15%. Infections occurred in 9.3% and 9.9%, respectively, of patients in the drug switch and reference filgrastim groups. Only 1 person in the drug switch group was hospitalized, which was a result of FN and occurred in cycle 6. Adverse events were similar between the drug switch and reference drug groups (42.1% and 39.2%, respectively). Bone pain occurred in 30.8% and 33.3% of the switch and reference groups, respectively.24
Filgrastim-sndz was compared with filgrastim for healthy donor stem-cell mobilization in a trial that compared reference filgrastim (N=107), filgrastim-sndz (N=85), and lenograstim (N=121).25 The study patients received 7.5 to 10 µg/kg daily, which was rounded to the nearest 300-µg or 480-µg syringe. The patients received G-CSF starting 4 days before stem cell collection. When comparing the 2 filgrastim drugs, the median patient weight was higher in the filgrastim-sndz group (82 vs 74 kg; P=.0262), but the reference filgrastim group had a higher body mass index (25.6 vs 24.2 kg/m2; P=.018). There was a slightly higher weight-based dose of filgrastim-sndz compared with that of filgrastim (9.8 vs 9.3 µg/kg; P<.001); however, the mean white blood cell count at mobilization was similar between the 2 arms (45.2 vs 46.4 g/L, respectively; P=.246). The mean CD34+ cells at apheresis were also similar between the 2 arms (119 vs 124 cells/uL; P=.354). This noninferiority was sustained even when correcting for sex, although the study investigators did not comment on the power of this trial. This trial did not assess the adverse-event differences between the filgrastim drugs.25
Last, tbo-filgrastim was compared with filgrastim in a prospective, randomized trial of patients undergoing mobilization for a first autologous HCT for multiple myeloma or non-Hodgkin lymphoma (NHL).26 A total of 46 patients were included in the tbo-filgrastim arm and 51 were included in the filgrastim arm. The patients received 10 µg/kg daily of each agent for 5 consecutive days. On the evening of day 4, every patient received plerixafor 0.24 mg/kg rounded to the nearest milligram. Patients could receive a second dose of plerixafor if they did not meet their collection goal on day 5. In all, 89% of the patients in this analysis received an autologous HCT for the treatment of multiple myeloma. Other than a slightly older population in the tbo-filgrastim group than in the filgrastim cohort (mean age, 62.4 vs 58 years; P=.014), there were no significant differences in the baseline characteristics of the 2 arms.26
In the primary end point of mean CD34+ yield on day 5 of mobilization, there was no significant difference between the tbo-filgrastim and filgrastim arms (11.6 vs 10 cells/kg; P=.873).26 Noninferiority was reached even on multivariate analysis through controlling for diagnosis, volume of blood processed, and age, although this showed a nonsignificant trend towards increased mobilization in the tbo-filgrastim arm (P=.214). There was no difference in the day-5 mean CD34+ cell mobilization, with 109.7 cells/µL in the tbo-filgrastim cohort and 92.1 cells/µL in the filgrastim cohort (P=.158). On day 4, before the administration of plerixafor, there was no difference in the mean CD34+cell mobilization between the tbo-filgrastim and filgrastim arms (26.2 vs 23.9 cells/uL, respectively; P=.604). No differences were seen between the 2 arms in neutrophil and platelet engraftments, with neutrophils engrafting in 11 days and platelets engrafting in 18 days in both arms. Bone pain occurred in 41% of patients in the tbo-filgrastim arm and in 43% of patients in the filgrastim arm (P=.855). Other adverse events occurred in similar frequencies in both arms, with no significant differences.26
Based on this information, trials have shown no difference between filgrastim and filgrastim biosimilars in infectious end points, including in the rates of FN and documented infections, although there was a difference in ANC at the time of FN. For cell mobilization, the biosimilars were similar with regards to white blood cell counts at mobilization and CD34+ counts, as well as in the final cells collected. Of note, not all of the trials discussed here were specifically noninferiority trials and some did not address power; however, the similarity in results of these trials to the noninferior trials is important to note. Data from the trials mentioned here on filgrastim and its biosimilars are shown in Appendix Table 1.23-26
Pegfilgrastim
Pegfilgrastim is a pegylated form of filgrastim that leads to an enhanced duration of action similar to filgrastim.18,27 Compared with filgrastim’s half-life of approximately 3.5 hours, pegfilgrastim’s half-life is from 15 to 80 hours in adults.18,27 The indications and adverse events for pegfilgrastim are similar to those for filgrastim, although because of its longer half-life pegfilgrastim requires less frequent dosing of 6 mg once per cycle of chemotherapy.27 The prescribing information recommends the avoidance of pegfilgrastim administration 14 days before the next cycle of chemotherapy.27 The FDA has approved 6 biosimilars to pegfilgrastim: pegfilgrastim-apgf (Nyvepria), pegfilgrastim-bmez (Ziextenzo), pegfilgrastim-cbqv (Udenyca), pegfilgrastim-fpgk (Stimufend), pegfilgrastim-jmdb (Fulphila), and pegfilgrastim-pbbk (Fylnetra).28-33
Pegfilgrastim-bmez was compared with pegfilgrastim in 2 separate trials (PROTECT-1 and PROTECT-2) as well as in a pooled analysis of the 2 trials.34-36 Both trials included patients with early-stage breast cancer who were receiving docetaxel, doxorubicin, and cyclophosphamide in the neoadjuvant or adjuvant setting. The pooled analysis included 314 patients who received pegfilgrastim-bmez and 310 who received pegfilgrastim. The baseline characteristics were well-balanced between the 2 arms, although the breakdown of patients receiving neoadjuvant and adjuvant chemotherapies was not reported in the trials. The dose intensities of docetaxel, doxorubicin, and cyclophosphamide were similar between the 2 arms.34-36
In the primary end point of mean duration of severe neutropenia, there was no significant difference between the biosimilar pegfilgrastim and pegfilgrastim arms (1.05 vs 1.01 days, respectively).34-36 This amounts to a difference of –0.04 days (95% CI, –0.19-0.11), which met the predefined noninferiority CI of ±1 day. The secondary end points were also similar between the 2 arms. The mean ANC nadirs were 0.8×109/L with pegfilgrastim-bmez and 0.687×109/L with pegfilgrastim, and the median time to ANC recovery in the first cycle was 1.84 days and 1.88 days, respectively. The adverse-event profiles of pegfilgrastim-bmez and pegfilgrastim were similar, with bone pain in 4.5% and 6.1% of the cohorts, respectively, and FN in 8% and 10% of patients, respectively.34-36 Data from the PROTECT-1 and PROTECT-2 trials are shown in Appendix Table 1.34,35
Last, pegfilgrastim-jmdb was compared with pegfilgrastim in a phase 3, randomized, double-blind study in patients with stage II or III breast cancer who were receiving docetaxel, doxorubicin, and cyclophosphamide in the neoadjuvant or adjuvant setting every 3 weeks for 6 cycles. G-CSF was given 24 hours after chemotherapy (plus a 2-hour window after the first 24 hours).37 A total of 127 patients received pegfilgrastim-jmdb and 67 patients received pegfilgrastim. No differences were noted in the baseline characteristics between the 2 arms. In all, 60% of the overall cohort received docetaxel, doxorubicin, and cyclophosphamide in the adjuvant setting. In the per-protocol population, the mean and median durations of severe neutropenia were similar in the pegfilgrastim-jmdb and pegfilgrastim arms (1.2 days and 1 day, respectively), which led to the 95% CI (–0.285-0.298) being within the prespecified noninferiority margin of ±1 day, which established equivalence. All secondary end points were equivalent between the biosimilar pegfilgrastim and reference pegfilgrastim arms, including the mean time to ANC nadir (6.2 vs 6.3 days, respectively), mean time to ANC recovery (1.9 vs 1.7 days, respectively), grade ≥3 neutropenia (91% vs 82%, respectively), rate of FN (6% vs 2%, respectively), and chemotherapy dose reductions or omissions (4% vs 2%, respectively). Bone pain occurred in similar rates between the 2 arms (40% vs 36%, respectively), although the investigators also state that naproxen was used for bone pain in the first cycle with the reference drug (28% vs 20%, respectively).37
Similar to filgrastim, the pegfilgrastim biosimilars are comparable with reference pegfilgrastim in infectious complications. There were no significant differences between the pegfilgrastim biosimilars and reference pegfilgrastim in the rate of neutropenia, FN, rate of hospitalizations, and dose reductions of chemotherapy secondary to neutropenia. Trial data on pegfilgrastim and one of its biosimilars are shown in Appendix Table 1.37
Rituximab
Rituximab was the first monoclonal antibody approved for use in patients with a hematology or oncology indication. Rituximab is a CD20-directed monoclonal antibody that activates complement-dependent and antibody-dependent cellular cytotoxicities.38 Rituximab is currently approved for the treatment of NHL, B-cell NHL, B-cell acute leukemia, chronic lymphocytic leukemia (CLL), and rheumatologic and autoimmune disease indications. Premedication is recommended with acetaminophen, an antihistamine, and a steroid; however, premedication can vary by institution. Infusion reactions can occur with rituximab treatment and can be fatal, although these reactions are most common after receiving the first dose of rituximab. Although rituximab is dosed at 375 mg/m2 in patients with NHL or CLL, patients with other indications may receive a flat dose, ie, a dose that is not based on weight. The frequencies and durations of dosing can vary based on the indication.38 Currently, there are 3 approved biosimilars for rituximab, including rituximab-abbs (Truxima), rituximab-arrx (Riabni), and rituximab-pvvr (Ruxience).39-41
The first rituximab biosimilar approved in the United States, rituximab-abbs, was assessed for efficacy in a phase 3 trial of patients with newly diagnosed follicular lymphoma (FL).42 Patients with low-grade FL per the Groupe D’Etude des Lymphomes Folliculaires criteria were randomized to rituximab-abbs (N=130) or reference rituximab (N=128) dosed at 375 mg/m2 weekly for 4 doses. The patients with controlled disease after 4 treatment cycles continued receiving the same agent every 8 weeks for 6 cycles. A second maintenance period could be completed if the patients completed the first maintenance period, in which rituximab-abbs was given to all patients at the investigator’s discretion.42
The baseline demographics were well-balanced between the 2 groups.42 Of the total patients, 77% completed all maintenance treatment cycles, including 97 patients who switched from reference rituximab to the rituximab biosimilar in the second maintenance treatment. Using central review, the ORRs were similar between the 2 arms, at 88% and 87% in the rituximab-abbs and rituximab arms, respectively; the CR rates were also similar, at 56% and 52%, respectively. Local review yielded similar results, and there was a high concordance between the local and central reviews. Because of the switch to the biosimilar in the second year of maintenance therapy, the investigators assessed for ORRs at the end; there were similar ORRs for the biosimilar and reference arms that switched (97% and 94%, respectively). The 24-month OS rate was 98% for both arms. The 24-month PFS rates were 88% and 83% for the biosimilar and reference drug arms, respectively. The adverse events were similar between the 2 arms. In all, 45% of patients who received rituximab-abbs and 41% of patients who received rituximab had infections, and infusion reactions occurred in 32% and 30% of the cohorts, respectively.42
The phase 3 JASMINE trial compared rituximab-arrx (N=123) with rituximab (N=124) in patients with grade I to grade IIIa asymptomatic FL.43 The patients received 375 mg/m2 of their respective agent weekly for 4 weeks followed by doses at weeks 12 and 20, with a final assessment at week 28. No significant between-group differences were noted in the patients’ baseline characteristics, although most patients in this study had low-risk classification per the Follicular Lymphoma International Prognostic Index (FLIPI; 44.1%) and did not have bone marrow involvement (70.7%). There was no difference in ORR by week 28 between the rituximab-arrx and rituximab arms (78% vs 70.2%, respectively; 95% CI, –3.2-18.6), which was within the a priory noninferiority margin of –15%. The CR rates were also similar in the rituximab-arrx and rituximab groups (23.6% vs 25.8%, respectively). Numerically, more patients in the rituximab-arrx arm had a partial response (54.5% vs 41.9%) and more patients in the reference arm had stable disease (28.2% vs 18.7%), although the investigators did not comment on the significance of these values. Infusion reactions occurred at similar rates in the rituximab-arrx and rituximab arms (43% vs 42.9%, respectively), as did the rate of overall adverse events (83.6% vs 75.4%, respectively).43
In a randomized, double-blind trial, the investigators compared rituximab-pvvr (N=196) with rituximab (N=198) in patients with treatment-naïve FL.44 The patients received rituximab-pvvr or rituximab at a dose of 375 mg/m2 once per week for 4 weeks and were then monitored for 1 year. No differences were noted in the patients’ baseline characteristics, although most (66%) patients did have moderate-risk disease per FLIPI2 scoring. The primary end point of ORR at week 26 was 75.5% in the rituximab-pvvr group and 70.7% in the rituximab group, for a difference of 4.66% (95% CI, –4.16-13.47%), which was within the predefined noninferiority margin of ±16% and thus established equivalence between the 2 arms. The CR rates were also similar in the rituximab-pvvr and rituximab arms (29.3% vs 31.%, respectively). The estimated 1-year PFS rate was 78.2% with rituximab-pvvr and 83% with rituximab (HR, 1.393; 95% CI, 0.847-2.291; P=.189). There was no significant difference in adverse events between the rituximab-pvvr and rituximab arms, with the most common adverse events being infusion reactions (25% vs 29.9%, respectively), pruritus (6.6% vs 11.2%, respectively), and headache (8.2% vs 9.6%, respectively). Grade ≥3 adverse events were also similar between the rituximab-pvvr and rituximab arms (13.8% vs 12.2%, respectively).44
As previously noted, in several trials rituximab biosimilars have shown noninferiority to reference rituximab primarily in the treatment of low-grade lymphoma. The ORRs, PFS rates, and DOR rates have been similar between rituximab and rituximab biosimilars. Data from the trials mentioned here on rituximab and its biosimilars are shown in Appendix Table 1.42-44
Trastuzumab
Trastuzumab is an anti-HER2 monoclonal antibody that binds to the extracellular domain of the HER2 protein.45 After binding, trastuzumab inhibits the proliferation of cancer by binding and mediating antibody-dependent cellular cytotoxicity. Trastuzumab is approved for the treatment of adjuvant or metastatic HER2-positive breast cancer and HER2-positive metastatic gastric and gastroesophageal junction cancer. Trastuzumab has 2 dose strategies, including weekly dosing with 4 mg/kg for the first dose and 2 mg/kg for subsequent doses, as well as every-3-week dosing with 8 mg/kg for the first dose and 6 mg/kg for subsequent doses. Trastuzumab has a Black Box warning for risks for embryo-fetal adverse events as well as infusion reactions, pulmonary adverse events, and cardiomyopathy, although cardiomyopathy is often subclinical. The prescribing information for trastuzumab recommends the avoidance of concurrent treatment with trastuzumab and anthracycline because of a compounded effect on left ventricular ejection fraction (LVEF) dysfunction.45 The FDA has approved 5 biosimilars for trastuzumab, including trastuzumab-anns (Kanjinti), trastuzumab-dkst (Ogivri), trastuzumab-dttb (Ontruzant), trastuzumab-pkrb (Herzuma), and trastuzumab-qyyp (Trazimera).46-50
Trastuzumab-anns was compared with trastuzumab in the phase 3 LILAC study in women with early-stage, HER2-positive breast cancer with plans for surgical resection.51 All patients received 4 cycles of neoadjuvant epirubicin 90 mg/m2 and cyclophosphamide 600 mg/m2 every 3 weeks for 4 cycles. After this, all patients received 4 cycles of paclitaxel 175 mg/m2 (or 80 mg/m2 if that was the center’s standard of care) and trastuzumab-anns or trastuzumab before having surgical resection. After receiving 4 cycles of epirubicin plus cyclophosphamide and paclitaxel with trastuzumab, patients then had surgery. In the adjuvant setting, patients could then continue a trastuzumab biosimilar (N=364) or reference trastuzumab (N=190), but a third group of patients who initially received trastuzumab in the adjuvant setting were switched to trastuzumab-anns (N=171).51
Regardless of the drug, all patients received trastuzumab for up to 1 year.51 Trastuzumab was administered at the standard dose of 8 mg/kg for the first dose and then 6 mg/kg for subsequent doses. The patients’ baseline characteristics, including relative dose intensity, were similar between all 3 arms. The pathologic complete response (pCR) rate after neoadjuvant therapy, which was the primary outcome in this analysis, was 48% in the trastuzumab-anns arm and 41% in the trastuzumab arm. By local review, this led to an RR of 1.188 (90% CI, 1.033-1.366), which crossed the upper limit of the equivalence margin of 1.318; however, on central review, the pCR rates were 48% with the trastuzumab biosimilar and 42% with reference trastuzumab (RR, 1.142; 90% CI, 0.993-1.312), which was within the noninferiority margin of 0.759 to 1.318. Despite local review crossing the upper margin, this was in favor of biosimilar trastuzumab-anns versus trastuzumab. No adverse-event differences were noted between the 2 arms in this analysis.51
A separate analysis of cardiac adverse events was conducted for the LILAC study to assess the long-term impact of trastuzumab-anns and trastuzumab on patients’ LVEF.52 LVEF decline was defined as a decrease from baseline of ≥10% to <50%. LVEF was measured using a 2-dimensional echocardiogram. Echocardiograms were performed at baseline and at the last visit during the neoadjuvant phase, as well as at cycles 9 and 13 and at the last cycle of the adjuvant treatment phase (ie, cycle 17). There were no significant differences in cardiac disorders at baseline in the biosimilar (17.6%), reference (15.3%), and drug switch (15.2%) groups. The median baseline rate of LVEF was approximately 65% in all arms. No differences were seen in LVEF decline between the 3 arms, and only 3.1% of the entire cohort had a decline in LVEF as defined above. All declines in LVEF were not clinically meaningful and were asymptomatic.52
The phase 3 HERITAGE trial compared trastuzumab-dkst with reference trastuzumab in patients with treatment-naïve metastatic breast cancer.53-55 All patients received an institution-preferred taxane and were randomized 1:1 to receive trastuzumab-dkst or trastuzumab. Treatment was received for a minimum of 8 cycles (24 weeks). If patients had at least stable disease at week 24, they were eligible to continue anti-HER2 therapy until disease progression, unacceptable adverse events, or death. The baseline characteristics were similar between the 2 arms. There was no significant difference in ORR between the trastuzumab-dkst and trastuzumab arms (69.6% vs 64%, respectively; RR, 1.09; 90% CI, 0.974-1.211), which was within the prespecified noninferiority margin of ±15%. The median PFS was 11.1 months in both arms. A subsequent analysis showed a correlation between ORR at week 24 and PFS in responders and nonresponders, with a biserial correlation coefficient of 0.752. The final OS results after 36 months also showed no difference in OS between the 2 groups. The median OS in the biosimilar arm and reference trastuzumab arm was 35 months and 30.2 months, respectively (HR, 0.87; 95% CI, 0.67-1.14; P=.325). There were no between-group differences in adverse events throughout the analysis. The investigators concluded that there were no meaningful differences in efficacy or adverse events between trastuzumab-dkst and reference trastuzumab, which indicates equivalence.53-55
In a phase 3, randomized, double-blind trial, trastuzumab-dttb was compared with trastuzumab in patients with early-stage HER2-positive breast cancer.56-58 All patients received 4 cycles of docetaxel followed by 4 cycles of fluorouracil, epirubicin, and cyclophosphamide concurrently with 8 total cycles of applicable trastuzumab or trastuzumab-dttb in the neoadjuvant setting. The patients then received the same HER2 therapy in the adjuvant setting for 10 cycles. A total of 380 patients received trastuzumab-dttb and 384 patients received trastuzumab. The baseline characteristics were similar between the 2 arms. In the primary end point of breast pCR, the rates were 51.7% and 42% in the trastuzumab-dttb and trastuzumab arms, respectively (adjusted ratio, 1.259; 95% CI, 1.085-1.46), which met the noninferiority margin of a 95% CI between 0.785 and 1.546 or a difference in breast pCR rates of ±13%. The adjusted difference in breast pCR was 10.7% (95% CI, 4.13-17.26), with the lower limit within the equivalence margin and the upper limit outside of the equivalence margin in favor of trastuzumab-dttb. The 4-year follow-up of this analysis showed event-free survival rates of 83.4% with trastuzumab-dttb and 80.7% with trastuzumab (HR, 0.77; 95% CI, 0.47-1.27) and 4-year OS rates of 94.3% and 89.6%, respectively (HR, 0.53; 95% CI, 0.24-1.16). Only 3 patients in this analysis had a decrease in LVEF (none of which were clinically significant), with all patients eventually recovering to an LVEF of ≥50%. All other adverse events were comparable between the groups and were expected with the agents in the trial.56-58
In a randomized, double-blind, phase 3 trial, trastuzumab-pkrb was compared with trastuzumab in patients with early-stage HER2-positive breast cancer.59-61 The patients received trastuzumab-pkrb (N=271) or trastuzumab (N=278) with concurrent fluorouracil, epirubicin, and cyclophosphamide for 8 cycles followed by surgery and adjuvant HER2 therapy. No significant differences were noted in the patients’ baseline characteristics. The primary end point of pCR was similar between the trastuzumab-pkrb and trastuzumab arms (46.8% vs 50.4%, respectively; 95% CI, –0.12-0.05), with a 95% CI within the prespecified equivalence margin of ±0.15. The median disease-free survival (DFS), PFS, and OS were not reached for either arm, and the HRs showed equivalence for all. The HRs were 1.23 (95% CI, 0.78-1.93) for DFS, 1.31 (95% CI, 0.86-2.01) for PFS, and 1.1 (95% CI, 0.57-2.13) for OS. Adverse events, including cardiac disorders, were similar among the groups. Cardiac disorders occurred in 11.8% of patients in the trastuzumab-pkrb group and in 14% of patients in the trastuzumab group.59-61
In a randomized, double-blind trial by Pegram and colleagues, trastuzumab-qyyp was assessed for clinical equivalence in 707 patients with treatment-naïve metastatic breast cancer.62 The patients received paclitaxel 80 mg/m2 on days 1, 8, and 15 of 28-day cycles with weekly trastuzumab-qyyp (N=352) or trastuzumab (N=355). Although the patients initially received weekly treatment, there was an option to change to the every-3-week dosing of trastuzumab starting at week 33. Paclitaxel was continued for at least 6 cycles or until maximum benefit was obtained, at which point patients continued to receive anti-HER2 monotherapy. The baseline characteristics were well-balanced between the arms, including estrogen receptor status; approximately 90% of the patients in both groups had not previously received trastuzumab.62
The ORRs were 62.5% and 66.5% in the trastuzumab-qyyp and trastuzumab arms, respectively (RR, 0.94; 95% CI, 0.842-1.049).62 Equivalence was met as a result of this CI meeting the prespecified noninferiority margin of 0.8 to 1.25. The best ORRs were similar between the trastuzumab-qyyp and trastuzumab arms, with the most frequent response being a partial response (59.7% and 62.8%, respectively).62 With longer follow-up, there was no difference in OS rates between the 2 arms.63 The 2-year and 3-year OS rates were 82.3% and 77.2%, respectively, for trastuzumab-qyyp and 77.4% and 75.3%, respectively, for trastuzumab (HR, 0.929; 95% CI, 0.656-1.316; P=.339). The adverse events were similar between the trastuzumab and biosimilar arm, including decreased ejection fraction (13.3% vs 14%, respectively).63 The study investigators concluded that there were no clinically relevant differences between the 2 arms.62,63
Trastuzumab and its biosimilars have shown noninferiority with one another in the previously noted trials, primarily in the end points of response rates (ie, pCR and ORR). In addition, all of the trastuzumab biosimilars except trastuzumab-anns have shown similar survival to that with trastuzumab in patients with breast cancer. Trastuzumab-anns was not assessed for survival. Trial data on trastuzumab and its biosimilars are shown in Appendix Table 1.51,53-63
Discussion
Biosimilars are often thought to have similar efficacy to the reference drug and are accepted in place of the reference drug, pursuant to state laws, by the American Society of Clinical Oncology (ASCO).64,65 The acquisition costs of biosimilars are also lower than for the reference drug, so there is also a considerable potential cost-savings for all involved in the healthcare process, from payer to patient.66 Despite this, none of the currently available biosimilars in oncology is considered interchangeable with the reference drug per the FDA, which requires 2 criteria to show interchangeability.65 The first criterion is for the biologic to show biosimilarity to the reference drug through pharmacokinetics and pharmacodynamic studies. The second criterion is for any biologic that is dosed more than once to show no difference in efficacy or adverse events when the new biosimilar is switched with the reference drug compared with receiving only the reference drug. These switch studies would need to show patient groups receiving the reference drug and the biosimilar in sequence and not have any new or increased adverse events.65
To date, all biosimilar comparison trials of approved biosimilars have shown at least noninferiority in efficacy and adverse events between the biosimilar and reference drug for the 5 currently available monoclonal antibodies with biosimilars (epoetin alfa was not approved at the time this article was written and is therefore not included).15-17,23-26,34-37,42-44,51-63 Several agents, including filgrastim-sndz, rituximab-abbs, and trastuzumab-anns, are no different from the reference drug in switch trials, indicating potential interchangeability status per the current FDA guidelines.24,42,51,65 A summary of the approved biosimilar comparison trials is shown in Appendix Table 1.
In addition to approved biosimilar agents, some trials have compared non–FDA-approved biosimilars to reference drugs.67-83 These trials have shown similar results to trials incorporating approved agents, but they are not discussed in detail here. A summary of trials that include these agents is shown in Appendix Table 2, available Here.67-83 Should these agents gain approval, the increased competition may further drive prices down.
Despite this evidence showing no apparent differences between biosimilars and reference drugs, there is a high misconception about their equivalence among oncologists.84 In a survey of 269 oncologists by Peipert and colleagues that was presented at the 2021 ASCO Quality Symposium, only approximately 66% of oncologists knew that biosimilars had a different chemical composition from the reference drug, and 23% to 30% had concerns about the efficacy and adverse events related to biosimilars. Interestingly, approximately 23% of the oncologists acknowledged having a lack of knowledge on biosimilars.84 Nearly all of the oncologists cited society guidelines, cost differences, adverse events, and efficacy as helping them to decide whether to adopt the use of biosimilars. Concerns about adverse events were a minority opinion (23% of the overall cohort), although physicians in private practice and community hospitals were more likely to share these concerns compared with physicians in academic hospitals (P=.02).84 Another analysis echoed these findings of a large variance in the knowledge of biosimilars, with the primary concern being adverse events.85 This analysis showed that physicians are more likely to prescribe biosimilars in treatment-naïve patients, which indicates a concern about switching between 2 drugs.85 It is important to note, however, that analyses such as this include nononcology specialties and are not reflective of a single physician population.
There is also concern among patients about switching biosimilars, as shown in a survey of 1696 patients in which 85% said that they would not want to switch drugs if their current drug was helping their disease.86 The survey respondents were concerned that the new drug would not treat their disease. In all, 83% of the patients were concerned that there would be new adverse events with the drug they switch to.86 In another survey of 3198 people, including patients in support groups, caregivers, and the general population, there was a lack of knowledge of biosimilars. Of the total survey respondents, 70% had never heard of biosimilars, although awareness was higher in patients (9%) and patients in a support group (20%) compared with the general population (6%; P<.05 between the support group and the other arms). There were differences in the perceptions of efficacy and adverse events as well, with 10% to 20% differences in the understanding of efficacy and adverse events compared with the reference and biosimilar drugs.87
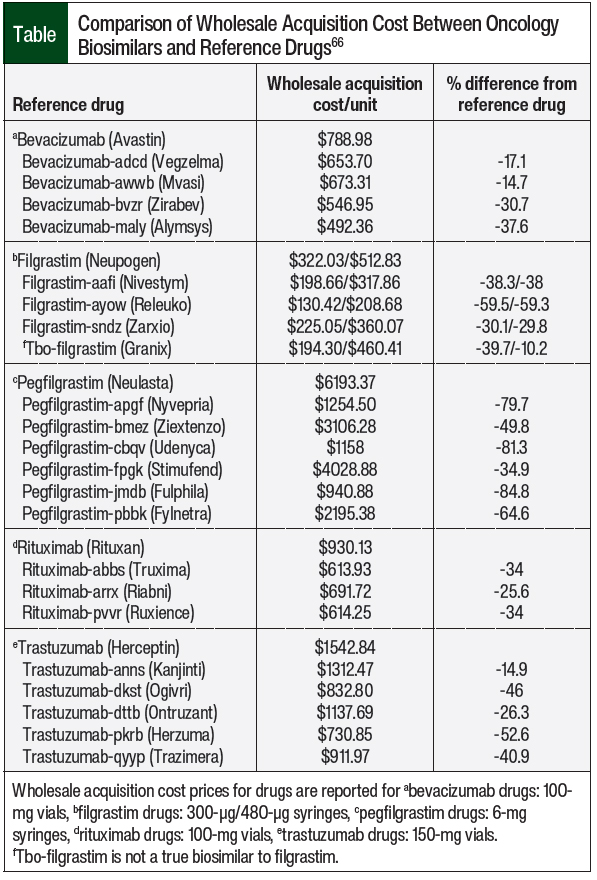
Despite these perceptions by oncologists and patients, the data presented here have shown that biosimilars are noninferior to reference drugs. The imperative to lower drug costs demands an increase in the availability of less-expensive drug alternatives, and the proliferation of biosimilars can fulfill this need. When assessing the wholesale acquisition costs (WACs) of biosimilars versus their reference drugs, they range between 13% and 30% less expensive for bevacizumab biosimilars, 10% to 53% less expensive for filgrastim biosimilars, 44% to 85% less expensive for pegfilgrastim biosimilars, 25% to 34% less expensive for rituximab biosimilars, and 15% to 53% less expensive for trastuzumab biosimilars (Table).66 Thus, to curb the costs of and increase the familiarity with biosimilars, increased utilization of these drugs is needed. As previously noted, commercial and public payers have implemented biosimilars into their formularies with significant cost-savings to the healthcare system.6-8 The Table includes a summary of WACs for the currently available oncology biosimilars.
Education is imperative in increasing familiarity with biosimilars among oncologists and patients.88,89 The adoption of biosimilars is feasible, and they are able to be incorporated quickly into health systems. The implementation of biosimilars through Pharmacy & Therapeutics (P&T)-approved pharmacy-driven substitutions can lead to the rapid adoption of a biosimilar within 12 months of protocol implementation.90 Education on how biosimilars relate to the reference drug has resulted in dramatic increases in the use of biosimilars, with one trial showing utilization increasing from 1% to 96% in less than 2 years, leading to subsequent cost-savings of $651,000 across the study period.91 Through pharmacist-led patient counseling, the use of biosimilars can be increased, with one study showing an increase from 41.1% to 69.4% after pharmacist-led counseling.92 Errors in prescribing can also be avoided; one survey showed that before education, approximately 20% of prescribers would consider switching a patient from a reference drug to a biosimilar if the patient was not responding to the reference drug.93 Pharmacists, as the medication specialists on the team, are uniquely positioned to clear up misunderstandings of biosimilars. Education on biosimilars for providers and patients can decrease uncertainty about these drugs in both groups and can ensure that they still trust that the medications being administered are working while maintaining cost-savings.
Conclusion
In conclusion, based on the available evidence, there is no discernible difference between the currently available oncology biosimilars and reference drugs with regards to efficacy and adverse events. Educating involved parties, including providers and patients, can alleviate concerns about efficacy and adverse events, which can ease the transition from reference drugs to biosimilars. The increased use of biosimilars is feasible with education and P&T-approved buy-in, which can lead to the rapid implementation of biosimilars and a subsequent decrease in cost to health systems.
Author Disclosure Statement
Dr Marjoncu has no conflicts of interest to report.
References
- García JJ, Raez LE, Rosas D. A narrative review of biosimilars: a continued journey from the scientific evidence to practice implementation. Transl Lung Cancer Res. 2020;9:2113-2119.
- Bell D. New FDA-approved oncology drugs (2021-2022). ASCO Post. June 3, 2022. https://ascopost.com/issues/june-3-2022-narratives-special-issue/new-fda-approved-oncology-drugs-2021-2022/. Accessed December 17, 2022.
- Kirchhoff CF, Wang XZM, Conlon HD, et al. Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng. 2017;114:2696-2705.
- US Food and Drug Administration. Implementation of the Biologics Price Competition and Innovation Act of 2009. February 12, 2016. www.fda.gov/drugs/guidance-compliance-regulatory-information/implementation-biologics-price-competition-and-innovation-act-2009. Accessed December 17, 2022.
- Hernandez I, Bott SW, Patel AS, et al. Pricing of monoclonal antibody therapies: higher if used for cancer? Am J Manag Care. 2018;24:109-112.
- Chambers JD, Lai RC, Margaretos NM, et al. Coverage for biosimilars vs reference products among US commercial health plans. JAMA. 2020;323:1972-1973.
- Karaca-Mandic P, Chang J, Go R, et al. Biosimilar filgrastim uptake and costs among commercially insured, Medicare Advantage. Health Aff (Millwood). 2019;38:1887-1892.
- Qian J. Uptake and cost of biosimilar filgrastim among Medicare and Medicaid populations in 2015-2018. J Manag Care Spec Pharm. 2021;27:660-666.
- Avastin (bevacizumab) injection, for intravenous use [prescribing information]. Genentech; September 2022. www.gene.com/download/pdf/avastin_prescribing.pdf. Accessed July 12, 2023.
- Vegzelma (bevacizumab-adcd) injection, for intravenous use [prescribing information]. Celltrion; February 2023. http://celltrionusa.com/data/file/product/final-labeling-text_202302.pdf. Accessed July 12, 2023.
- Mvasi (bevacizumab-awwb) injection, for intravenous use [prescribing information]. Amgen; February 2023. www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/mvasi/mvasi_pi_hcp_english.pdf. Accessed July 12, 2023.
- Zirabev (bevacizumab-bvzr) injection, for intravenous use [prescribing information]. Pfizer; February 2023. https://labeling.pfizer.com/ShowLabeling.aspx?id=11860. Accessed July 12, 2023.
- Alymsys (bevacizumab-maly) injection, for intravenous use [prescribing information]. Amneal Pharmaceuticals; April 2022. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=6b4040a2-a5c5-4ff0-ab45-935d7e49cf78&type=display. Accessed July 12, 2023.
- US Food and Drug Administration. Biosimilar product information. www.fda.gov/drugs/biosimilars/biosimilar-product-information. Accessed December 18, 2022.
- Verschraegen C, Andric Z, Moiseenko F, et al. Candidate bevacizumab biosimilar CT-P16 versus European Union reference bevacizumab in patients with metastatic or recurrent non-small cell lung cancer: a randomized controlled trial. BioDrugs. 2022;36:749-760.
- Thatcher N, Goldschmidt JH, Thomas M, et al. Efficacy and safety of the biosimilar ABP 215 compared with bevacizumab in patients with advanced nonsquamous non-small cell lung cancer (MAPLE): a randomized, double-blind, phase III study. Clin Cancer Res. 2019;25:2088-2095. Erratum in: Clin Cancer Res. 2019;25:3193.
- Trukhin D, Poddubskaya E, Andric Z, et al; for the STELLA investigators. Efficacy, safety and immunogenicity of MB02 (bevacizumab biosimilar) versus reference bevacizumab in advanced non-small cell lung cancer: a randomized, double-blind, phase III study (STELLA). BioDrugs. 2021;35:429-444.
- Neupogen (filgrastim) injection, for subcutaneous or intravenous use [prescribing information]. Amgen; April 2023. www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/Neupogen/neupogen_pi_hcp_english.pdf. Accessed July 12, 2023.
- Nivestym (filgrastim-aafi) injection, for subcutaneous or intravenous use [prescribing information]. Pfizer; March 2023. https://labeling.pfizer.com/ShowLabeling.aspx?id=10899. Accessed July 12, 2023.
- Releuko (filgrastim-ayow) injection, for subcutaneous or intravenous use [prescribing information]. Kashiv BioSciences; Amneal Pharmaceuticals; April 2022. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=74e1ec6e-1630-4654-895c-2bd355f939e7&type=display. Accessed July 12, 2023.
- Zarxio (filgrastim-sndz) injection, for subcutaneous or intravenous use [prescribing information]. Sandoz; Novartis; September 2022. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=c0d1c22b-566b-4776-bdbf-00f96dad0cae&type=display. Accessed July 12, 2023.
- Granix (tbo-filgrastim) injection, for subcutaneous use [prescribing information]. Teva Pharmaceutical Industries; November 2019. www.granixhcp.com/globalassets/granix-hcp/prescribing-information.pdf. Accessed July 12, 2023.
- Brito M, Esteves S, André R, et al. Comparison of effectiveness of biosimilar filgrastim (Nivestim), reference Amgen filgrastim and pegfilgrastim in febrile neutropenia primary prevention in breast cancer patients treated with neo(adjuvant) TAC: a non-interventional cohort study. Support Care Cancer. 2016;24:597-603.
- Blackwell K, Gascon P, Krendyukov A, et al. Safety and efficacy of alternating treatment with EP2006, a filgrastim biosimilar, and reference filgrastim: a phase III, randomised, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2018;29:244-249.
- Farhan R, Urbanowska E, Zborowska H, et al. Biosimilar G-CSF versus filgrastim and lenograstim in healthy unrelated volunteer hematopoietic stem cell donors. Ann Hematol. 2017;96:1735-1739.
- Bhamidipati PK, Fiala MA, Grossman BJ, et al. Results of a prospective randomized, open-label, noninferiority study of tbo-filgrastim (Granix) versus filgrastim (Neupogen) in combination with plerixafor for autologous stem cell mobilization in patients with multiple myeloma and non-Hodgkin lymphoma. Biol Blood Marrow Transplant. 2017;23:2065-2069.
- Neulasta (pegfilgrastim) injection, for subcutaneous use [prescribing information]. Amgen; February 2021. www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/Neulasta/neulasta_pi_hcp_english.pdf. Accessed July 12, 2023.
- Nyvepria (pegfilgrastim-apgf) injection, for subcutaneous use [prescribing information]. Pfizer; March 2023. https://labeling.pfizer.com/ShowLabeling.aspx?format=PDF&id=13622. Accessed July 12, 2023.
- Ziextenzo (pegfilgrastim-bmez) injection, for subcutaneous use [prescribing information]. Sandoz; March 2021. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=7dada041-6528-4acf-809c-62d271538c9a&type=display. Accessed July 12, 2023.
- Udenyca (pegfilgrastim-cbqv) injection, for subcutaneous use [prescribing information]. Coherus BioSciences; March 2023. https://udenyca.com/hcp/wp-content/pdfs/udenyca-pi.pdf. Accessed July 12, 2023.
- Stimufend (pegfilgrastim-fpgk) injection, for subcutaneous use [prescribing information]. Fresenius Kabi USA; September 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2022/761173Orig1s000correctedlbl.pdf. Accessed July 12, 2023.
- Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use [prescribing information]. Mylan Institutional; October 2021. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=3ea915d7-2feb-4e75-91f7-913c965b7d8a&type=display. Accessed July 12, 2023.
- Fylnetra (pegfilgrastim-pbbk) injection, for subcutaneous use [prescribing information]. Kashiv BioSciences; Amneal Pharmaceuticals; May 2022. www.accessdata.fda.gov/drugsatfda_docs/label/2022/761084s000lbl.pdf. Accessed July 12, 2023.
- Harbeck N, Lipatov O, Frolova M, et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol. 2016;12:1359-1367.
- Blackwell K, Donskih R, Jones CM, et al. A comparison of proposed biosimilar LA-EP2006 and reference pegfilgrastim for the prevention of neutropenia in patients with early-stage breast cancer receiving myelosuppressive adjuvant or neoadjuvant chemotherapy: Pegfilgrastim Randomized Oncology (Supportive Care) Trial to Evaluate Comparative Treatment (PROTECT-2), a phase III, randomized, double-blind trial. Oncologist. 2016;21:789-794.
- Blackwell K, Gascon P, Jones CM, et al. Pooled analysis of two randomized, double-blind trials comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol. 2017;28:2272-2277.
- Waller CF, Ranganna GM, Pennella EJ, et al. Randomized phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H in the prophylactic treatment of chemotherapy-induced neutropenia. Ann Hematol. 2019;98:1217-1224.
- Rituxan (rituximab) injection, for intravenous use [prescribing information]. Biogen; Genentech; December 2021. www.gene.com/download/pdf/rituxan_prescribing.pdf. Accessed July 17, 2023.
- Truxima (rituximab-abbs) injection, for intravenous use [prescribing information]. Celltrion; Teva Pharmaceuticals USA; February 2022. www.truximahcp.com/globalassets/truxima-dtc/pdfs/truxima-prescribing-information.pdf. Accessed July 17, 2023.
- Riabni (rituximab-arrx) injection, for intravenous use [prescribing information]. Amgen; June 2022. www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/Riabni/riabni_pi_english.pdf. Accessed July 17, 2023.
- Ruxience (rituximab-pvvr) injection, for intravenous use [prescribing information]. Pfizer; November 2021. https://labeling.pfizer.com/ShowLabeling.aspx?id=12090. Accessed July 17, 2023.
- Kwak LW, Sancho JM, Cho SG, et al. Efficacy and safety of CT-P10 versus rituximab in untreated low-tumor-burden follicular lymphoma: final results of a randomized phase III study. Clin Lymphoma Myeloma Leuk. 2022;22:89-97.
- Niederwieser D, Hamm C, Cobb P, et al. Efficacy and safety of ABP 798: results from the JASMINE trial in patients with follicular lymphoma in comparison with rituximab reference product. Target Oncol. 2020;15:599-611. Erratum in: Target Oncol. 2020;15:807.
- Sharman JP, Liberati AM, Ishizawa K, et al. A randomized, double-blind, efficacy and safety study of PF-05280586 (a rituximab biosimilar) compared with rituximab reference product (MabThera) in subjects with previously untreated CD20-positive, low-tumor-burden follicular lymphoma (LTB-FL). BioDrugs. 2020;34:171-181.
- Herceptin (trastuzumab) for injection, for intravenous infusion [prescribing information]. Genentech; February 2021. www.gene.com/download/pdf/herceptin_prescribing.pdf. Accessed July 17, 2023.
- Kanjinti (trastuzumab-anns) for injection, for intravenous use [prescribing information]. Amgen; October 2022. www.pi.amgen.com/-/media/Project/Amgen/Repository/pi-amgen-com/kanjinti/kanjinti_pi.ashx. Accessed July 27, 2023.
- Ogivri (trastuzumab-dkst) for injection, for intravenous use [prescribing information]. Mylan Institutional, a Viatris Company; February 2021. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?type=display&setid=6b7938e6-14c7-4a65-9605-967542ecfb8f. Accessed July 27, 2023.
- Ontruzant (trastuzumab-dttb) for injection, for intravenous use [prescribing information]. Organon; June 2021. www.organon.com/product/usa/pi_circulars/o/ontruzant/Ontruzant-pi.pdf. Accessed July 27, 2023.
- Herzuma (trastuzumab-pkrb) for injection, for intravenous use [prescribing information]. Celltrion; Teva Pharmaceuticals USA; May 2019. www.accessdata.fda.gov/drugsatfda_docs/label/2019/761091s001s002lbl.pdf. Accessed July 27, 2023.
- Trazimera (trastuzumab-qyyp) for injection, for intravenous use [prescribing information]. Pfizer; December 2020. https://labeling.pfizer.com/ShowLabeling.aspx?id=12725. Accessed July 27, 2023.
- von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19:987-998.
- Kolberg HC, Colleoni M, Demetriou GS, et al. Cardiac safety of the trastuzumab biosimilar ABP 980 in women with HER2-positive early breast cancer in the randomized, double-blind, active-controlled LILAC study. Drug Saf. 2020;43:233-242.
- Rugo HS, Barve A, Waller CF, et al; for the Heritage Study investigators. Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317:37-47.
- Rugo HS, Pennella EJ, Gopalakrishnan U, et al. Correlation between week 24 trastuzumab-dkst response and week 48 progression-free survival: the HERITAGE trial. Breast. 2021;58:18-26.
- Rugo HS, Pennella EJ, Gopalakrishnan U, et al. Final overall survival analysis of the phase 3 HERITAGE study demonstrates equivalence of trastuzumab-dkst to trastuzumab in HER2-positive metastatic breast cancer. Breast Cancer Res Treat. 2021;188:369-377.
- Pivot X, Bondarenko I, Nowecki Z, et al. Phase III, randomized, double-blind study comparing the efficacy, safety, and immunogenicity of SB3 (trastuzumab biosimilar) and reference trastuzumab in patients treated with neoadjuvant therapy for human epidermal growth factor receptor 2–positive early breast cancer. J Clin Oncol. 2018;36:968-974.
- Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
- Pivot X, Pegram MD, Cortes J, et al. Four-year follow-up of a phase III study comparing SB3 (trastuzumab biosimilar) and reference trastuzumab in HER2-positive early or locally advanced breast cancer in neoadjuvant setting. J Clin Oncol. 2020;38(15 suppl):Abstract 578. 2020 ASCO Annual Meeting I.
- Stebbing J, Baranau Y, Baryash V, et al. CT-P6 compared with reference trastuzumab for HER2-positive breast cancer: a randomised, double-blind, active-controlled, phase 3 equivalence trial. Lancet Oncol. 2017;18:917-928. Errata in: Lancet Oncol. 2017;18:e433; Lancet Oncol. 2017;18:e510.
- Esteva FJ, Baranau YV, Baryash V, et al. Efficacy and safety of CT-P6 versus reference trastuzumab in HER2-positive early breast cancer: updated results of a randomised phase 3 trial. Cancer Chemother Pharmacol. 2019;84:839-847.
- Stebbing J, Baranau YV, Baryash V, et al. Long-term efficacy and safety of CT-P6 versus trastuzumab in patients with HER2-positive early breast cancer: final results from a randomized phase III trial. Breast Cancer Res Treat. 2021;188:631-640.
- Pegram MD, Bondarenko I, Moreira Costa Zorzetto M, et al. PF-05280014 (a trastuzumab biosimilar) plus paclitaxel compared with reference trastuzumab plus paclitaxel for HER2-positive metastatic breast cancer: a randomised, double-blind study. Br J Cancer. 2019;120:172-182.
- Li RK, Tokunaga E, Adamchuk H, et al. Long-term safety and effectiveness of PF-05280014 (a trastuzumab biosimilar) treatment in patients with HER2-positive metastatic breast cancer: updated results of a randomized, double-blind study. BioDrugs. 2022;36:55-69.
- Nahleh Z, Lyman GH, Schilsky RL, et al. Use of biosimilar medications in oncology. JCO Oncol Pract. 2022;18:177-186.
- US Food and Drug Administration. Considerations in Demonstrating Interchangeability With a Reference Product: Guidance for Industry. May 2019. www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-product-guidance-industry. Accessed December 22, 2022.
- Merative. Micromedex RED BOOK. June 6, 2023. www.merative.com/clinical-decision-support. Accessed July 27, 2023.
- Syrigos K, Abert I, Andric Z, et al; for the AVANA investigators. Efficacy and safety of bevacizumab biosimilar FKB238 versus originator bevacizumab: results from AVANA, a phase III trial in patients with non-squamous non-small-cell lung cancer (non-sq-NSCLC). BioDrugs. 2021;35:417-428.
- Qin S, Li J, Bai Y, et al. Efficacy, safety, and immunogenicity of HLX04 versus reference bevacizumab in combination with XELOX or mFOLFOX6 as first-line treatment for metastatic colorectal cancer: results of a randomized, double-blind phase III study. BioDrugs. 2021;35:445-458.
- Shi Y, Lei K, Jia Y, et al. Bevacizumab biosimilar LY01008 compared with bevacizumab (Avastin) as first-line treatment for Chinese patients with unresectable, metastatic, or recurrent non-squamous non–small-cell lung cancer: a multicenter, randomized, double-blinded, phase III trial. Cancer Commun (Lond). 2021;41:889-903.
- Hegg R, Mattar A, Nunes de Matos-Neto J, et al. A phase III, randomized, non-inferiority study comparing the efficacy and safety of biosimilar filgrastim versus originator filgrastim for chemotherapy-induced neutropenia in breast cancer patients. Clinics (Sao Paulo). 2016;71:586-592.
- Sivgin S, Karakus E, Keklik M, et al. Evaluation of the efficacy and safety of original filgrastim (Neupogen), biosimilar filgrastim (Leucostim) and lenograstim (Granocyte) in CD34+ peripheral hematopoietic stem cell mobilization procedures for allogeneic hematopoietic stem cell transplant donors. Transfus Apher Sci. 2016;54:410-415.
- Engert A, Griskevicius L, Zyuzgin Y, et al. XM02, the first granulocyte colony-stimulating factor biosimilar, is safe and effective in reducing the duration of severe neutropenia and incidence of febrile neutropenia in patients with non-Hodgkin lymphoma receiving chemotherapy. Leuk Lymphoma. 2009;50:374-379.
- Kowalyszyn RD, Fein LE, Richardet ME, et al. Biosimilar versus originator pegfilgrastim for preventing chemotherapy-induced neutropenia: a phase III randomized, multicenter, evaluator-blinded, noninferiority study. JCO Glob Oncol. 2022;8:e2100276.
- Kahan Z, Grecea D, Smakal M, et al. Efficacy and safety of RGB-02, a pegfilgrastim biosimilar to prevent chemotherapy-induced neutropenia: results of a randomized, double-blind phase III clinical study vs. reference pegfilgrastim in patients with breast cancer receiving chemotherapy. BMC Cancer. 2019;19:122.
- Poddubnaya IV, Alekseev SM, Kaplanov KD, et al. Proposed rituximab biosimilar BCD-020 versus reference rituximab for treatment of patients with indolent non-Hodgkin lymphomas: an international multicenter randomized trial. Hematol Oncol. 2020;38:67-73.
- Jurczak W, Moreira I, Kanakasetty GB, et al. Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol. 2017;4:e350-e361.
- Shi Y, Song Y, Qin Y, et al. A phase 3 study of rituximab biosimilar HLX01 in patients with diffuse large B-cell lymphoma. J Hematol Oncol. 2020;13:38.
- Song Y, Zhou H, Zhang H, et al. Efficacy and safety of the biosimilar IBI301 plus standard CHOP (I-CHOP) in comparison with rituximab plus CHOP (R-CHOP) in patients with previously untreated diffuse large B-cell lymphoma (DLBCL): a randomized, double-blind, parallel-group, phase 3 trial. Adv Ther. 2021;38:1889-1903.
- Candelaria M, González DE, Torresan Delamain M, et al; for the RTXM83 study. Rituximab biosimilar RTXM83 versus reference rituximab in combination with CHOP as first-line treatment for diffuse large B-cell lymphoma: a randomized, double-blind study. Leuk Lymphoma. 2019;60:3375-3385.
- Alexeev SM, Khorinko AV, Mukhametshina GZ, et al. Randomized double-blind clinical trial comparing safety and efficacy of the biosimilar BCD-022 with reference trastuzumab. BMC Cancer. 2020;20:783.
- Pivot X, Georgievich MA, Shamrai V, et al. Efficacy of HD201 vs referent trastuzumab in patients with ERBB2-positive breast cancer treated in the neoadjuvant setting: a multicenter phase 3 randomized clinical trial. JAMA Oncol. 2022;8:698-705. Erratum in: JAMA Oncol. 2022;8:784.
- Xu B, Zhang Q, Sun T, et al. Efficacy, safety, and immunogenicity of HLX02 compared with reference trastuzumab in patients with recurrent or metastatic HER2-positive breast cancer: a randomized phase III equivalence trial. BioDrugs. 2021;35:337-350.
- Nodehi RS, Kalantari B, Raafat J, et al. A randomized, double-blind, phase III, non-inferiority clinical trial comparing the efficacy and safety of TA4415V (a proposed trastuzumab biosimilar) and Herceptin (trastuzumab reference product) in HER2-positive early-stage breast cancer patients. BMC Pharmacol Toxicol. 2022;23:57.
- Peipert J, Kaiser K, Mehta Kircher S, et al. Oncologists’ knowledge and perspectives on the use of biosimilars. J Clin Oncol. 2021;39(28 suppl):Abstract 35. 2021 ASCO Quality Care Symposium.
- Sarnola K, Merikoski M, Jyrkkä J, Hämeen-Anttila K. Physicians’ perceptions of the uptake of biosimilars: a systematic review. BMJ Open. 2020;10:e034183.
- Teeple A, Ginsburg S, Howard L, et al. Patient attitudes about non-medical switching to biosimilars: results from an online patient survey in the United States. Curr Med Res Opin. 2019;35:603-609.
- Jacobs I, Singh E, Sewell KL, et al. Patient attitudes and understanding about biosimilars: an international cross-sectional survey. Patient Prefer Adherence. 2016;10:937-948.
- Leonard E, Wascovich M, Oskouei S, et al. Factors affecting health care provider knowledge and acceptance of biosimilar medicines: a systematic review. J Manag Care Spec Pharm. 2019;25:102-112.
- Tadjalli Oskouei S, Kusmierczyk AR. Biosimilar uptake: the importance of healthcare provider education. Pharmaceut Med. 2021;35:215-224.
- Waterhouse DM, Burdette C, Davies D, et al. Scaled integration of FDA approved biosimilars: closing the knowledge and adoption gaps. J Clin Oncol. 2021;39(28 suppl):Abstract 15. 2021 ASCO Quality Care Symposium.
- Morris GA, McNicol M, Boyle B, et al. Increasing biosimilar utilization at a pediatric inflammatory bowel disease center and associated cost savings: show me the money. Inflamm Bowel Dis. 2022;28:531-538.
- Levivien C, Bottois C, López Medina C, et al. Impact of a clinical pharmacist in a multidisciplinary consultation on the switch to a biosimilar for inflammatory rheumatic diseases. Joint Bone Spine. 2022;89:105322.
- Gibofsky A, Badawi S. Biosimilar knowledge among US rheumatologists – a survey. Arthritis Rheumatol. 2017;69(suppl 10):Abstract 1037. ACR/ARHP Annual Meeting Abstract Supplement.