CON: Katherine Mandock, PharmD, BCPS; Scott A. Soefje, PharmD, BCOP
PRO: Val R. Adams, PharmD, BCOP, FCCP
The Case Against Appoval
Controversy has developed over the label indications for bevacizumab in metastatic breast cancer (MBC). The US Food and Drug Administration (FDA) had granted accelerated approval for bevacizumab for the treatment of MBC based on clinical trials that demonstrated a progression-free survival (PFS) advantage. This controversy begins when the postapproval trials required with the FDA approval failed to demonstrate an overall survival (OS) advantage in addition to the PFS advantage. Subsequently, the Oncology Drug Advisory Committee (ODAC) recommended, and ultimately the FDA issued its decision, to remove bevacizumab’s current labeled indication for MBC.1
In this debate we take the side that supports the FDA’s final decision to remove that indication. Our position is based on the arguments that reflect the regulatory, efficacy, safety, and economic perspectives. We will demonstrate that this indication does not meet the standard required for labeled indications, that the efficacy for the indication does not show a clinical benefit, that there are serious safety concerns, and that the economic impact is too great to justify the drug’s approval.
The FDA Modernization Act of 1997 permitted the FDA to approve the marketing of drugs “upon a determination that the product has an effect on a clinical endpoint or on a surrogate endpoint that is reasonably likely to predict clinical benefit.”2 This is exactly what has happened with bevacizumab.
However, the FDA’s Guidance for Industry also states, “Where an accelerated approval is based upon a surrogate endpoint or on an effect on a clinical endpoint other than survival or irreversible morbidity, postmarketing studies are ordinarily required ‘to verify and describe the drug’s clinical benefit and to resolve remaining uncertainty as to the relation of the surrogate endpoint upon which approval was based to clinical benefit, or the observed clinical benefit to ultimate outcome (57 FR 58942, December 11, 1992).’”3 This is the focus of the regulatory argument.
The FDA has not recognized PFS as an end point that will grant approval for first-line indications in oncology. PFS has not translated to OS in any clinical trial in MBC and is, therefore, not considered a surrogate for survival. The burden is then on the drug manufacturer to demonstrate a clinical benefit that meets the criteria for approval, for example, OS, improved quality of life, or another end point that the FDA has recognized.
One other such example in oncology is gefitinib, which was approved on a surrogate end point and was then pulled from the market when the primary end point was not met. Bevacizumab has not met the requirements for full approval for an MBC indication; therefore, the FDA’s decision was the correct one.
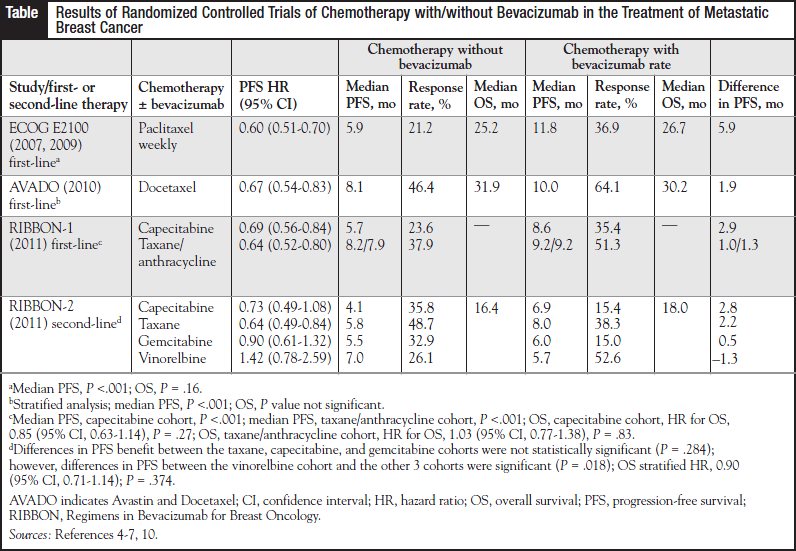
The results of the Eastern Cooperative Oncology Group (ECOG) E2100 trial formed the basis for the FDA’s decision to grant bevacizumab conditional accelerated approval for the first-line treatment of patients with MBC. The ECOG E2100 study demonstrated an almost 2-fold increase in response rate and time to progression, but it failed to show OS benefit when bevacizumab was added to weekly paclitaxel therapy.4,5
Two additional phase 3 trials were designed to validate the results from the E2100 study, while evaluating the use of bevacizumab in combination with other approved chemotherapy in the first-line treatment of MBC.6,7
The AVADO (Avastin and Docetaxel) trial examined the combination of docetaxel and bevacizumab administered every 3 weeks6; RIBBON (Regimens in Bevacizumab for Breast Oncology)-1 assessed cape - citabine, a taxane-based regimen (docetaxel or nabpaclitaxel) administered every 3 weeks, and an anthracycline- containing regimen administered alone or in combination with bevacizumab.7
View larger version
As in the case of the E2100 trial, both AVADO and RIBBON-1 failed to demonstrate a significant difference in OS rates. Both trials did demonstrate statistically significant improvements in PFS and in the response rate; however, they failed to confirm the magnitude of the PFS benefit observed in the E2100 trial (Table). Statistical significance does not always correspond to clinical significance, and the clinical impact of adding bevacizumab to a regimen is unconvincing at best, in view of the incremental improvements in PFS observed in the confirmatory trials.6,7
The concern for the clinical benefit of bevacizumab as a first-line agent in MBC is multifactorial but hinges on 3 major issues: (1) the disparity of benefit observed between the E2100 trial and other phase 3 trials, (2) the suitability of PFS as a surrogate end point for OS, and (3) the lack of a clearly defined patient population that is most likely to benefit from the addition of bevacizumab to the treatment regimen. The disparity in the magnitude of improvement in PFS between E2100 and other phase 3 studies is most likely reflective of the choice of chemotherapy.
The E2100 trial evaluated bevacizumab in combination with once-weekly paclitaxel therapy. Preclinical evidence suggests an antiangiogenic activity that is associated with weekly paclitaxel therapy, which, when combined with bevacizumab, may have acted synergistically to result in the greater duration of PFS observed in E2100. Therefore, the robust benefit in PFS that was seen with E2100 was most likely derived from fractionating the dose of paclitaxel weekly over 3 weeks rather than from the addition of bevacizumab to the regimen.8
In addition, all 3 studies used PFS as the primary efficacy end point.6-8 As noted earlier, PFS has not been demonstrated as a good surrogate for OS in solid tumors, including MBC.9
Finally, the collective randomized controlled trials have failed to identify a patient population that would derive the greatest potential benefit from the addition of bevacizumab to chemotherapy. Heavily pretreated patients are not likely to experience significant benefits with the addition of bevacizumab, as seen by the shorter PFS among the capecitabine-receiving cohort in RIBBON-1 versus patients who received taxane or anthracycline chemotherapy.7
An earlier trial comparing the efficacy of capecitabine with or without bevacizumab showed no differences in OS or PFS, confirming that bevacizumab is not likely to have a significant survival impact in patients with MBC who have received extensive treatment.10
In addition, data from RIBBON-2 suggest that in some chemotherapy cohorts, combinations with bevacizumab may have a negative impact on PFS.11 In a subgroup analysis of second-line agents, the vinorelbine cohort demonstrated shorter PFS and OS compared with the placebo arm. These results must be interpreted with caution, however, because they are based on a small sample size that included a greater percentage of patients with poor prognostic factors.11 In addition to questions regarding its clinical significance, the combination of bevacizumab and chemotherapy raises some serious safety concerns. After convergent safety results from the phase 3 trials, the ATHENA (Avastin Therapy for Advanced Breast Cancer) trial was conducted to further assess the safety of bevacizumab combined with first-line chemotherapy.12 The most frequent grade 3 or more toxicities in ATHENA were neutropenia (5.4%), hypertension (4.4%), thromboembolism (3.2%), proteinuria (1.7%), and bleeding (1.4%), which confirmed the previously established adverse events (AEs) associated with bevacizumab therapy.12
RIBBON-1 demonstrated that hypertension and proteinuria were consistently increased in the bevacizumab arms, regardless of the chemotherapy administered. In addition, the incidence of bleeding and febrile neutro - penia was >5% in patients receiving taxane-containing regimens.7 Disconcertingly, it appears that the more serious AEs occur more frequently in the population of patients most likely to benefit from bevacizumab’s favorable effects on PFS, namely, patients receiving weekly paclitaxel therapy.7
With no evidence of improved survival in phase 3 clinical trials, bevacizumab has failed to meet the conditions set under the current FDA regulatory standards for approval of first-line treatment for MBC, and it is associated with potentially serious AEs. Until further studies confirm an OS benefit or clinically and statistically significant improvements in PFS in addition to an ideal target population, bevacizumab should not be considered as a first-line agent in combination with chemotherapy for MBC.
From an economics perspective, the use of bevacizumab in MBC is not cost-effective. The estimated cost for 1 year of life saved with a course of bevacizumab is $496,000.13 Compared with conventional values between $50,000 and $125,000 for other therapies, this estimated cost of bevacizumab is several times higher. The cost of bevacizumab is approximately $62,000 in the clinical trials.13 The cost of the drug does not seem out of line with the cost of current cancer therapies; however, the benefit—in this case, years of life saved—is so small that the cost-effective amount, which is cost divided by years of life saved, becomes very large and outside the range that is generally acceptable.
In these times of medical economic scarcity, the continued approval of a drug that shows marginal benefit is not in society’s best interest. If asked to pay out of pocket, how many patients would accept bevacizumab, knowing its high cost and the minimal additional life gained?
We have demonstrated that bevacizumab has not overcome the regulatory or efficacy hurdles required for approval of a first-line agent for MBC. Combined with the safety concerns and the tremendous associated cost, the FDA’s decision to pull the indication for the drug in the treatment of MBC is acceptable.
THE CASE FOR CONTINUED APPROVAL
Based on the scientific data and the regulatory requests, it is clear that the MBC indication should remain on the label of bevacizumab. The rationale for continued approval is based on the arguments related to the 4 areas cited earlier: regulatory, efficacy, safety, and economic.
As noted before, the Modernization Act permits the FDA to approve a drug based on a surrogate end point that is reasonably likely to predict clinical benefit.2 This is the way the first-line indication for paclitaxel and bevacizumab for MBC was approved. The pivotal study for the accelerated approval for bevacizumab is the ECOG E2100 trial.4 This trial randomized 722 patients to paclitaxel alone or to paclitaxel plus bevacizumab. The trial was designed to measure PFS as the primary end point; it required 546 events (in 685 patients) to detect a 2-month improvement, with a power of 85%. The improvement in PFS was impressive—5.8 months without paclitaxel alone versus 11.3 months for paclitaxel plus bevacizumab—and led to the FDA’s accelerated approval. The secondary end points were also promising, an improved response rate of 37% versus 21% (P <.001), and improved 1-year survival rate of 81% versus 73% (P = .01), respectively.4 As is the case with all accelerated approvals, the FDA requires ≥1 postmarketing studies to confirm or refute the perceived benefit that had led to the accelerated approval.3
Because of the business/financial risk involved with performing large trials that may be rejected by the FDA, the process proceeds with planning meetings that include the FDA, to ensure that the required end points and trial design are clearly defined and deemed acceptable. Accordingly, during a meeting between the manufacturer (Genentech) and the FDA (a type B meeting) on February 2, 2009, the “FDA confirmed that the basis for conversion to full approval will be demonstrated improvement in progression-free survival and evidence that survival is not impaired.”14 This is the key focus of the regulatory argument.
With this confirmation, Genentech carried out the RIBBON-1 and the AVADO trials, which were designed to demonstrate (1) a benefit in PFS, and (2) no detriment to OS. The postmarketing studies presented to the FDA included the E2100 (N = 722), RIBBON-1 (N = 1237), and AVADO (N = 736) trials, which totaled >2500 patients randomized and analyzed in these trials.4-7,10 The combined data with 24-month updated data from AVADO showed median 1.9 months and mean 2.5 months of improvement in PFS, which were statistically significant (hazard ratio, 0.67). This clearly meets the first requirement of the FDA.14 The 1-year OS was 77% and 82%, respectively, with a projected median survival of 26.4 and 26.7 months, respectively, with chemotherapy alone or with bevacizumab. Although not superior, this clearly demonstrates that survival was not impaired by treatment with bevacizumab.14
The primary concern of ODAC and of the FDA that led to the removal of the approval stemmed from the survival data (ie, evidence that patients lived longer) and the quality-of-life data.15 This is unfortunate, because the trials were neither designed nor powered to answer the survival question. With the first evaluation of the E2100 study, one can substitute the survival numbers for event rate to see that the study is underpowered to answer the OS question.4,5 The trial’s statistical design stated the need for 546 PFS events in 685 patients to reach the 2-month threshold, with a power of 85%; however, only 483 deaths occurred among the 722 patients, with a projected survival difference of 1.7 months.4 This is close to the unwritten 2-month survival advantage that will reset the standard for diseases such as lung cancer.16,17
One of the reasons it is unfortunate that the E2100 trial was not designed or powered to answer the OS question is that the study was done before the drug’s approval, which minimized the use of second-line bevacizumab. The RIBBON-1 study was designed with 2 cohorts: (1) a capecitabine ± bevacizumab cohort, and (2) a taxane/anthracycline ± bevacizumab cohort.7 The capecitabine cohort required 405 PFS events for 600 patients to detect a 2-month survival difference; again, substituting deaths for PFS events makes it easy to see that the study was underpowered; only 191 deaths occurred among the 605 patients.7 The crossover factor further confounds the OS data: 85% of the placeboreceiving patients were given ≥1 subsequent chemo - therapy regimens, and >50% of those patients received chemotherapy plus bevacizumab.7 Therefore, comparing OS in this study would actually compare the timing (first line vs second line) rather than the true impact of bevacizumab versus no bevacizumab.
In the end, it is clear that the postmarketing studies conducted to facilitate the conversion of the initial accelerated approval to the final regular FDA approval were not designed, powered, or intended to answer the impact on OS. Instead, they were designed, carried out, and proved improved PFS, without deleterious effects on survival, which had been the FDA’s agreed upon end point for conversion to regular approval.14 From a regulatory perspective, therefore, bevacizumab clearly should be approved for breast cancer, which would require the FDA to honor its commitment and advice regarding required regulatory end points.
The key question in this debate relates to efficacy—Is a 2.5-month improvement in PFS and increased response rate sufficient for the agent to be used (off label) as first-line treatment for MBC? To answer this question in a more objective fashion, we need to consider other precedence in MBC, such as the addition of capecitabine to docetaxel (compared with docetaxel alone). The pivotal study for approval of this combination regimen came from a single study with a combined 511 patients in the 2 arms (255/256).18
The results from this trial are very similar to the data seen with bevacizumab plus chemotherapy: a 58-day increase in PFS, an increase from 22% to 32% in response rate, and a 90-day increased survival. The chemotherapy plus bevacizumab improvements in PFS and response rate are numerically better than these data. Although we do not know the true effect on survival (as discussed above), if we estimate the impact to be approximately 2 months, this would seem to reach the usual survival threshold.
Consequently, I would propose that we could compare the efficacy of bevacizumab and chemotherapy to the capecitabine plus docetaxel regimen. Because the utilization of capecitabine and docetaxel varies, this perhaps could be used as a litmus test: if you use docetaxel and capecitabine, then you should consider using bevacizumab plus chemotherapy; if you do not believe that adding capecitabine to docetaxel provides enough benefit to warrant using this combination, you would likely not consider using bevacizumab plus chemotherapy.
In summary, I would argue that the threshold for efficacy has been determined by other regimens (primarily by capecitabine added to docetaxel), and that bevacizumab added to chemotherapy appears to provide very similar results and should therefore be available for use in this setting.
The other side of efficacy is the toxicity level, or safety, that is required to ensure a net benefit. It is clear that efficacy trumps toxicity; this is most clearly demonstrated with allogeneic hematopoietic stem-cell transplant (SCT) for acute myeloid leukemia (AML).19 However, when the efficacy is more marginal than the results with allogeneic SCT in AML, safety concerns become more importantly and appropriately scrutinized.
The pooled data presented to the FDA for bevacizumab clearly show increased overall rates of grade 3 and 4 toxicity with the addition of bevacizumab compared with chemotherapy alone (23% vs 36%, respectively). 14 The toxicities in the combination group were known, expected, and consistent with adding bevacizumab to chemotherapy in other settings. The most common safety issues included increased rates of hypertension and proteinuria (approximately 11% increased rate with bevacizumab), as well as an increase in arterial thromboembolism and bleeding (approximately 1% higher rate for each).14
Again, it is important to compare this to previous acceptable evidence, such as in the capecitabine and docetaxel regimen in MBC. The pivotal study for capecitabine shows that the combination of these agents led to at least 1 grade 3 toxicity in 76.5% of patients compared with 57.6% of patients with docetaxel alone.18 Although the toxicity level is different between capecitabine and bevacizumab, adding capecitabine to docetaxel increased the percentage of patients who had at least 1 grade 3 AE more than the addition of bevacizumab to chemotherapy (19% vs 13%, respectively). This is an appropriate comparison for toxicity, because as stated above, the efficacy is also similar.
In addition, it is important to note that regimens that generate grade 3 and 4 AEs in the range of a 36% rate are common; this reminds us that preventing and managing these toxicities is crucial. Careful monitoring of patients’ blood pressure level and urine for protein, with appropriate intervention, can easily minimize these bevacizumab-induced additional toxicities.14
In summary, the increased toxicity is real and quantifiable; however, it is easy to monitor and manage, and it certainly does not meet a threshold that would restrict the drug from being used on or off label.
The last consideration is economics, which arguably is the driving force behind this debate. By removing the approval of bevacizumab for MBC, insurance companies and the Centers for Medicare & Medicare Services have a reason to deny payment for this expensive therapy. The cost of therapy, however, is not under the purview of the FDA’s authority and should not impact approval or disapproval of a drug. In a real-world discussion regarding economics, not paying for bevacizumab for patients with breast cancer because of its high cost would seem unreasonable at many levels.
An objective way to evaluate the drug’s cost should consider other uses and benefits of bevacizumab, at similar doses. In the ECOG E3200 trial, patients with colorectal cancer received FOLFOX4 (5-fluorouracil, leucovorin, oxaliplatin) ± bevacizumab with a dose (10 mg/kg every 2 weeks) used in the E2100 breast cancer trial.20 The results of this trial showed that adding bevacizumab to this chemotherapy regimen increased PFS by 2.6 months, response rates by 14%, and OS by 2.1 months.20 Similarly, the ECOG 4599 trial evaluated carboplatin and paclitaxel ± bevacizumab (15 mg/kg every 3 weeks) in patients with advanced-stage non–small-cell lung cancer (NSCLC). The results of this study show that adding bevacizumab to this chemotherapy regimen increased PFS by 1.7 months, increased response rate by 20%, and OS by 2 months.17
These numbers look very similar to the cumulative results of adding bevacizumab (10 mg/kg every 2 weeks, or 15 mg/kg every 3 weeks) to chemotherapy for breast cancer: a PFS increase of 1.9 or 2.5 months (median and mean, respectively), increase in response rate of 11% to 26%, and a projected survival from the E2100 study (as discussed above) of 1.7 months. If one views the above data as similar (as I do), then saying that it is cost-prohibitive could be interpreted as saying that less money should be spent for patients with breast cancer than for those with colorectal or lung cancer. Such a conclusion, I believe, is ethically inappropriate.
In summary, although bevacizumab for MBC is expensive, its efficacy, dosing, and, therefore, cost are similar to those seen with the use of this drug in colorectal or lung cancer, which makes this argument defunct. Furthermore, the FDA does not consider cost in its drug approval decisions.
Finally, Genentech followed the regulatory process and sought guidance from the FDA to design trials and reach end points required to convert a conditional, accelerated approval to a full, final FDA approval. The cumulative data from these trials (with >2500 patients) demonstrate the achievement of PFS (median, 1.9 months; mean, 2.5 months) and no detrimental effect on survival. Regretfully, the FDA does not have a PFS threshold that the agency would consider approvable. This appears to be a “bait and switch” maneuver; the FDA advised or agreed to a PFS end point, never stating how much was enough, but in the end only voiced concern that there was no evidence to demonstrate a survival advantage.
The FDA made a motion to maintain its accelerated approval, while allowing appropriate OS data to be collected; and yet, the FDA decided to remove its initial approval. Based on the E2100 OS data (still inappropriate, but the best we have), the compiled PFS, safety data, and cost, bevacizumab should maintain its original approval, because the data look very similar to existing precedence in MBC (capecitabine added to docetaxel), colorectal cancer (bevacizumab added to FOLFOX4), and NSCLC (bevacizumab added to carboplatin plus paclitaxel).
References
- Pollack A. FDA revokes approval of Avastin for use as breast cancer drug. New York Times. November 18, 2011. www.nytimes.com/2011/11/19/business/fda-revokesapproval-of-avastin-as-breast-cancer-drug.html?_r=1&ref=health. Accessed November 20, 2011.
- US Food and Drug Administration Modernization Act of 1997, S 830, 105th Cong, 1st Sess (1997). www.fda.gov/downloads/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/SignificantAmendmentstotheFDCAct/FDAMA/FullTextofFDAMAlaw/UCM089145.pdf. Accessed November 20, 2011.
- US Food and Drug Administration. Guidance for industry: fast track drug development programs—designation, development, and application review. Procedural revision 2. January 2006. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079736.pdf. Accessed November 20, 2011.
- Miller K, Wang M, Grawlow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666-2676.
- Gray R, Bhattacharya S, Bowden C, et al. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2009;27:4966-4972. Epub 2009 Aug 31.
- Miles D, Chan A, Dirix LY, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2–negative metastatic breast cancer. J Clin Oncol. 2010;28:3239- 3247. Epub 2010 May 24.
- Robert NJ, Dieras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebocontrolled, phase III trial of chemotherapy with or without bevacizumab for first line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252-1260. Epub 2011 Mar 7.
- Seidman AD, Berry D, Cirrincione C, et al. Randomized phase III trial of weekly compared with every-3-week paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors: final results of the Cancer and Leukemia Group B Protocol 9840. J Clin Oncol. 2008;26:1642-1649.
- Burzykowski T, Buyse M, Piccart-Gebhart MJ, et al. Evaluation of tumor response, disease control, progression-free survival, and time to progression as potential surrogate end points in metastatic breast cancer. J Clin Oncol. 2008;26:1987-1992.
- Miller KD, Chap LL, Holmes FA, et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792-799.
- Brufsky AM, Hurvitz S, Perez E, et al. RIBBON-2: a randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of bevacizumab in combination with chemotherapy for second-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2011;29:4286-4293. Epub 2011 Oct 11.
- Smith IE, Pierga JY, Biganzoli L, et al, for the ATHENA Study Group. First-line bevacizumab plus taxane-based chemotherapy for locally recurrent or metastatic breast cancer: safety and efficacy in an open-label study in 2,251 patients. Ann Oncol. 2011;22:595-602. Epub 2010 Sep 5.
- Fojo T, Parkinson DR. Biologically targeted cancer therapy and marginal benefits: are we making too much of too little or are we achieving too little by giving too much? Clin Cancer Res. 2010;16:5972-5980.
- Horning S. Avastin combined with chemotherapy in HER2-negative first line metastatic breast cancer (MBC). www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM219979.pdf. Accessed November 30, 2011.
- National Cancer Institute. FDA approval for bevacizumab: metastatic HER2-negative breast cancer. November 18, 2011. www.cancer.gov/cancertopics/druginfo/fdabevacizumab. Accessed November 30, 2011.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): non-small cell lung cancer v.2.2012. Updated 2011. www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed November 30, 2011.
- Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non–small-cell lung cancer. N Engl J Med. 2006;355:2542-2550.
- Xeloda (capecitabine) package insert. Roche Pharmaceuticals; Nutley, NJ; 2011.
- Bishop MR, Pavletic SZ. Hematopoietic stem cell transplantation (Chapter 32). In: Abeloff MD, Armitage JO, Niederhuber JE, et al, eds. Abeloff’s Clinical Oncology. 4th ed. Philadelphia, PA: Churchill Livingston; 2008:501-512.
- Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539-1544.